
Department of Bioengineering, Integral University, Lucknow-26026, India
ABSTRACT: This study assessed the molecular interactions of (±)-anti-and-syndibenzo[a,l]pyrene-11,12-diol-13,14-epoxide (DBPDE), 7,12- dimethylbenz[a]anthracene-3,4-diol-1,2-epoxide (DMBADE), N2- hydroxylated-PhIP (N2-OH-PhIP), (±)-anti-and-syn-benzo[a]pyrene-7,8-diol9,10-epoxide (BPDE) with various Glutathione S-transferase (GST) and Nacetyltransferase (NAT) isozymes. Our in-silico data revealed that GSTP1 (- 8.83 kcal/mol), showing more plausible binding as compared to GSTM1 (-8.74 kcal/mol) and GSTA1 (ΔG: -8.03 kcal/mol) against (-)-anti-DBPDE and (+)- syn-DBPDE. We also investigated the involvement of GST and NAT isozymes in the conjugation of DMBADE and N2-OH-PhIP as a control despite their preferred routes sulfonation and glucuronidation for detoxification. The findings exhibited feeble binding of different classes of GSTs with metabolites of DMBA and PhIP, as highlighted by their free energy of binding. The enzymatic activity of GSTM1 against the most potent diol-epoxide of benzo[a]pyrene (BP), (+)-anti-BPDE, and (+)-syn-BPDE followed by GSTP1 and GSTA1 has well documented. In addition, these findings provide new perspectives for most probable mechanistic details of the detoxification pathway of PAHs and xenobiotics useful in combination therapy for future ligand-based drug discovery and development.
Keywords: DBP, DMBA, GST, NAT, Molecular, docking, AutoDock, Tools
Polycyclic aromatic hydrocarbons (PAHs) are widespread mutagenic and carcinogenic pollutants[1],[2] that require metabolic activation to electrophilic intermediates[3]-[5] and subsequent covalent adduct formation with cellular DNA to elicit their biological activity.[6],[7] However, several different activation pathways exhibited strong evidence of a prominent role of the bay-and-fjord region diol-epoxides as ultimate mutagenic [8],[9] and carcinogenic metabolites of PAHs.[3],[9] Diol-epoxides of PAHs have produced stereoselectivity in a consecutive sequence of reactions catalyzed by CYP isozymes (3) and microsomal epoxide hydrolase.[10] Principally they exist as two enantiomeric pairs of diastereomers designated (-) diol-epoxide-2 or (-)- anti-diol-epoxide, (+) diol-epoxide-2 or (+)-antidiol-epoxide, (-) diol-epoxide-1 or (-)-syn-diolepoxide, (+) diol-epoxide-1 or (+)-syn-diolepoxide.
The balance between the metabolic activation and detoxification process influences the intracellular level of diol-epoxide-DNA-adduct formation. Sterically hindered bay-region diol-epoxides show an apparent low affinity for microsomal epoxide hydrolase[11] and their half-lives, substantially increased by a stabilization effect of intracellular lipids.[12] At the same time, the spontaneous enzyme(s)-catalyzed hydrolysis to less toxic tetraols appear to ΔG of minor importance in the cellular defense against diol-epoxides appear to ΔG the glutathione S-transferase (GST) catalyzed conjugation with glutathione.[13]
Several studies indicate that the efficiency of inactivation by the pattern and the level of expressed GST isozymes, and the cellular level of glutathione.[14],[15] The conjugation of diastereomeric diol-epoxides of various PAHs was studied well with purified rodent human GSTs.[16],[17] It shows significant differences in the activity and enantioselectivity towards these metabolites, dependent on the absolute configuration and the geometry of the aromatic structure. For example, the stereoisomeric bay-region diol-epoxides, particularly the syn-diastereomers, were in most cases efficiently conjugated by human GST of class mu (GSTM) with wide variations in enantioselectivity ranging from 50-90%. In contrast, in most cases, human GST of class mu showed only an appreciable activity towards bayregion anti-diol-epoxides and a high preference for conjugation of enantiomers with (R, S, S, R)- configuration.[17]
Apart from GST isozymes, another vital phase II enzyme, N-acetyltransferase (NAT), plays an essential role in detoxifying heterocyclic amines and other xenobiotics.[18] These NAT isozymes can detoxify molecules by N-acetylation. They can also activate other molecules through O-acetylation to form reactive acetoxy-esters that are unstable and decompose to electrophiles that bind to DNA leading to mutagenesis and carcinogenesis.[18] Polymorphisms in NAT enzymes result in differences in acetylation rate (slow and rapid) and can occur more commonly in specific ethnic populations.[19] The GLY191ALA mutant in NAT2 is more common in the African population, but the GLY268GLU common in Asians. Slow acetylation rates may make individuals more susceptible to certain diseases, and numerous studies indicate a relationship between NAT polymorphisms and certain kinds of cancers.[20] ,[21] Slow NAT2 acetylation phenotypes have shown a relationship to urinary bladder cancer[22], while associated colon, breast, lung, and prostate cancers are more controversial.[23]
In present study we have comprehensively investigated and elucidated the mechanism of metabolic detoxification of diastereomers of dibenzo[a,l]pyrene (DBP) namely (-) DBPDE-2 or (- )-anti-DBPDE, (+) DBPDE-2 or (+)-anti-DBPDE, (-) DBPDE-1 or (-)-syn-DBPDE and (+) DBPDE-1 or (+)-syn-DBPDE (Figure 1); 7,12-DMBA-3,4-diol1,2-epoxide (DMBADE) of 7,12- dimethylbenz[a]anthracene (DMBA) (Figure 2); N2-hydroxylated metabolite (N2-OH-PhIP) of 2- amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) (Figure 3); (-) BPDE-2 or (-)-anti-BPDE, (+) BPDE-2 or (+)-anti-BPDE, (-) BPDE-1 or (-)-synBPDE and (+) BPDE-1 or (+)-syn-BPDE metabolites of benzo[a]pyrene (BP) (Figure 4) by various GST isozymes including GSTA1, A2, A3, A4, A5, GSTM1, M2, M3, M4, M5, GSTP1 and NAT isozymes including NAT1and NAT2 using computational approach.
Retrieval and optimization of proteins 3D structures
3D structures of GSTA1 (PDB: 1GSE), GSTA2 (1AGS), GSTA3 (1TDI), GSTA4 (1GUL), GSTM1 (2VCT), GSTM2 (1HNA), GSTM3 (3GTU), GSTM4 (4GTU), GSTP1 (13GS), NAT1 (2IJA) and NAT2 (2PFR) were retrieved from Protein Data Bank (http://www.rcsb.org/pdb), while 3D structures of GSTM5 and GSTA5 accessed from the SMR database (http://swissmodel.expasy.org/repository/). Proteins were prepared for docking by removing undesired atoms, ions, and molecules. By applying appropriate force field energy minimization, remove the bad steric clashes using the steepest descent algorithm for 1000 steps at an RMS gradient of 0.01. [24]
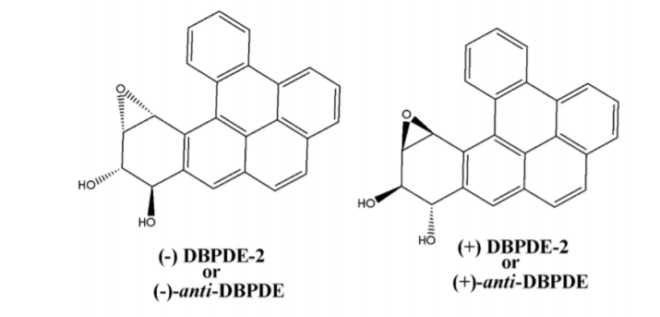
Figure 1: Chemical structure and absolute stereochemistry of anti-and syn-diol-epoxides of DBP.
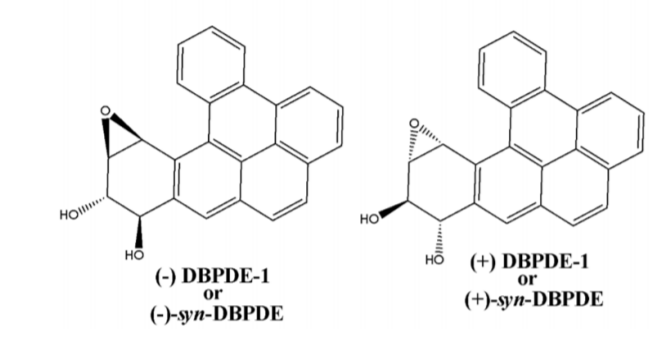
Figure 2: Chemical structure of DMBADE.
Figure 3: Chemical structure of N2-OH-PhIP.


Figure 4: Absolute stereochemistry of anti-and syn-diol-epoxides of BP.
Retrieval and optimization of ligands 3D structures
The molecular formula and SMILES notations of (±)-anti-DBPDE, (±)-syn-DBPDE, DMBADE, N2-OHPhIP, (±)-anti-BPDE and (±)-syn-BPDE retrieved from PubChem database (http://pubchem.ncbi.nlm.nih.gov) The SMILES string was taken for each of the ligands and submitted to another online software system CORINA (http://www.molecularnetworks.com/products/corina). It takes SMILES string as input and generates the 3D structure of the molecule for which structural coordinates file downloaded in PDB format suitable for AutoDock Tools 4.0.25 Appropriate force field was applied to them and subjected to single-step energy minimization using the steepest descent algorithm for 500 steps at an RMS gradient of 0.01.[24]
Active site identification
We identified the preferable ligand binding site of different GST and NAT isozymes by Q-Site finder (http://www.modelling.leeds.ac.uk/qsitefinder). It takes PDB files or PDB ID of proteins as input, predicts binding sites, and gives residues responsible for making a particular binding site along with a graphic view of that particular binding site as output. One of the active sites having a larger volume carried forward for docking to show molecular interactions with different ligands.[26]
Docking simulation
Docking studies of (±)-anti-DBPDE, (±)-syn-DBPDE, DMBADE, N2- OH-PhIP, (±)-anti-BPDE and (±)-synBPDE performed using the AutoDock Tools 4.0 to find the preferred binding conformations of the ligands in the receptor.[27] Docking to macromolecule was performed using an empirical free energy function and Lamarckian Genetic Algorithm, with an initial population of 250 randomly placed individuals, a maximum number of 106 energy evaluations, a mutation rate of 0.02, and a crossover rate of 0.80. One hundred independent docking runs executed for each ligand. Results differing by <2.0 Å in positional root-meansquare deviation (RMSD) were clustered together and represented by the result with the most favorable free energy of binding.[25]
Visualization of docked complexes
Ligplot: (http://biochem.ucl.ac.uk/bsm/ligplot/ligplot.htm l), a command-line-based program and ghost script viewer (http://www.cs.wisc.edu/~ghost/gsview/) was used to show the protein-ligand interactions. After successful execution, Ligplot generates a postscript file that further converted to desirable graphic images with the help of GSV.[28]
Validation of docking methodology
To validate the docking methodology, we re-docked the ligand present within the active site of GSTP1 (PDB: 13GS) using the same parameters used for docking the metabolites of DBP, DMBA, PhIP, and BP into its active site. The top-ranking conformations clustered in terms of root mean square deviation (RMSD) between docked position and experimentally determined position for the ligand.
Metabolic detoxification of (±)-anti-DBPDE, (±)- syn-DBPDE with GST and NAT isozymes
To validate the docking methodology, we re-docked the ligand present within the active site of GSTP1 (PDB: 13GS) using the same parameters used for docking the metabolites of DBP, DMBA, PhIP, and BP into its active site. The top-ranking conformations clustered in terms of root mean square deviation (RMSD) between docked position and experimentally determined position for the ligand.
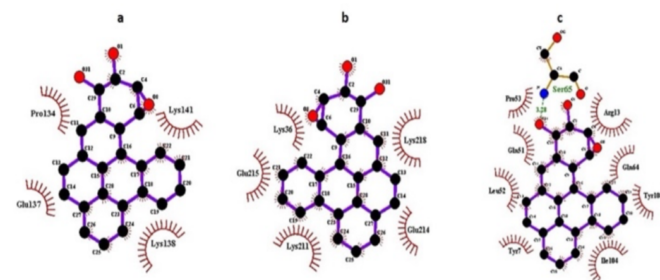
Figure 5: Ligplot showing molecular interactions of (-)-anti-DPBDE with a) GSTA1, b) GSTM1, c) GSTP1. Dashed lines and arcs show hydrophobic contacts and H-bonds, respectively.
Docking simulation exhibited the preferred binding pattern of (±)-anti-DBPDE and (±)-syn-DBPDE with various GST and NAT isozymes using AutoDock Tools 4.0. Our docking results revealed that (-)- anti-DPBDE showed a strong binding affinity with GSTP1 (ΔG: -8.83 kcal/mol, Ki: 0.338 µM) followed by GSTM1 (ΔG: -8.74 kcal/mol, Ki: 0.393 µM) and GSTA1 (ΔG: -8.03 kcal/mol, Ki: 1.30 µM). Furthermore, GLN51, SER65, TYR108, ILE04, ARG13, PRO53 residues of GSTP1, LYS36, GLU215, LYS211, GLU214 residues of GSTM1, PRO134, GLU137, LYS141, and LYS138 residues of GSTA1 (Figure 5a-c) are involved in hydrophobic interaction of (-)-anti-DPBDE. The binding pattern of GSTA and GSTM isozymes against the (-)-antiDPBDE were followed the order asGSTA1>A3>A4>A2>A5, GSTM1>M3>M5>M4> M2 (Table 1). In addition, our docking findings also revealed that NAT2 (ΔG: -9.37 kcal/mol, Ki: 0.135 µM) showed more favorable binding than NAT1 (ΔG: -5.84 kcal/mol, Ki: 52.84 µM) with (-)-antiDPBDE (Table 1). THR289, PHE37, SER125, GLY126, PHE217, LEU288, CYS68, PHE93, VAL106,ILE95 residues of NAT2 are hydrophobically involved with (-)-anti-DPBDE.
Further, (+)-anti-DPBDE showed slightly less binding affinity and different order as-GSTA1 (ΔG: – 6.44 kcal/mol, Ki: 18.90 µM) > GSTM1 (ΔG: -6.41 kcal/mol, Ki: 20.14 µM) > GSTP1 (ΔG: -6.37 kcal/mol, Ki: 21.56 µM) when compared to (-)-antiDPBDE. LYS141, LYS138, GLU137 residues of GSTA1, ALA38, GLU39, LYS218, GLU214 residues of GSTM1, TYR49, GLN51, GLN64, and ARG13 residues of GSTP1 are engaged in hydrophobic interaction with (+)-anti-DPBDE. The binding pattern of GSTA and GSTM isozymes against the (+)-anti-DPBDE were followed the order asGSTA1>A2>A4>A5>A3; GSTM1>M2>M4>M3> M5 (Table 1). In addition, our in-silico results also revealed that NAT2 (ΔG: -8.31 kcal/mol, Ki: 0.813 µM) showed more favorable binding than NAT1 (ΔG: -7.91 kcal/mol, Ki: 1.59 µM) with (+)-antiDPBDE (Table 1). THR289, LEU288, ILE95, VAL106, PHE93, PHE37, and GLY126 residues of NAT2 are hydrophobically involved (+)-antiDPBDE.
Our in-silico findings further elicited that (+)-synDPBDE showed a more favorable binding affinity with GSTM1 (ΔG: -8.36 kcal/mol, Ki: 0.749 µM) followed by GSTP1 (ΔG: -7.72 kcal/mol, Ki: 2.20 µM) and GSTA1 (ΔG: -6.89 kcal/mol, Ki: 7.55 µM). LYS218, ALA38, ARG221, PHE222 residues of GSTM1, ILE104, CYS101, ARG13, GLN51, GLN64 residues GSTP1, SER142, LYS141, LYS138, and GLY137 residues of GSTA1 are involved in hydrophobic interaction of (+)-syn-DPBDE. Furthermore, the binding pattern of GSTA and GSTM isozymes against the (+)-syn-DPBDE were followed the order as- GSTA1 >A4 >A3 >A5 >A2, GSTM1>M5>M4>M3>M2 (Table 1). In addition, our in-silico results also revealed that NAT2 (ΔG: – 9.71 kcal/mol, Ki: 0.075 µM) showed more favorable binding than NAT1 (ΔG: -6.25 kcal/mol, Ki: 26.15 µM) with (+)-syn-DPBDE (Table 1). THR289, LEU288, ILE95, VAL106, PHE93, PHE37, CYS68, SER125, and GLY126 residues of NAT2 are hydrophobically involved with (+)-syn-DPBDE.In continuation (-)-syn-DPBDE showed slightly less binding affinity and different order as- GSTM2 (ΔG: -6.01 kcal/mol, Ki: 39.70 µM) > GSTA4 (ΔG: -5.34 kcal/mol, Ki: 122.26 µM) > GSTP1 (ΔG: -4.55 kcal/mol, Ki: 461.55 µM) when compared to (+)- syn-DPBDE (Table 1) The binding pattern of GSTA and GSTM isozymes against the (-)-syn-DBPDE were followed the order asGSTA4>A5>A2>A3>A1, GSTM2>M3>M4>M1>M5 (Table 1). In addition, our docking findings also publicized that NAT2 (ΔG: -7.01 kcal/mol, Ki: 7.31µM) showed more favorable binding than NAT1 (ΔG: -6.90 kcal/mol, Ki: 8.83 µM) with (-)- syn-DPBDE (Table 1). THR289, LEU288, ILE95, VAL106, PHE93, PHE37, SER125, and GLY126 residues of NAT2 are engaged in hydrophobic interactions with (+)-syn-DPBDE.

Table 1: Molecular docking studies of (±)-diolepoxides of DBP with GST and NAT isozymes.
Most of the wet-lab data exhibited better enzymatic activity of GSTA1 against the most potent diolepoxide of DBP, namely (-)-anti-DBPDE and (+)- syn-DBPDE followed by GSTM1 and GSTP1 (GSTA1>GSTM1>GSTP1).[17],[29]-[31], However, our insilico findings showed the lowest Ki-in GSTP1 followed by GSTM1 and GSTA1 against (-)-antiDBPDE. In (+)-syn-DBPDE, GSTM1 showed the highest affinity, followed by GSTP1 and GSTA1. These results are not in complete harmony with wet-lab data that emphasized the significant enzymatic activity of GSTA1.[13],[17] Although there are no significant differences reflected in the above insilico findings, which means GSTA1, GSTM1, or GSTP1 might conjugate with (-)-anti-DBPDE. Furthermore, GSTA1, A2, A3, A4, and A5 (A1>A3>A4>A2>A5) also showed plausible binding activity with (-)-anti-DBPDE, which is nearly close to experimental data.[32] Further, in-silico results also revealed the significant involvement of GSTM1 (ΔG: -8.36 kcal/mol, Ki: 0.749 µM) followed by GSTP1 (ΔG: -7.72 kcal/mol, Ki: 2.20 µM) and GSTA1 (ΔG: -6.89 kcal/mol, Ki: 7.55 µM) towards the molecular interactions with other potent carcinogenic metabolites, (+)-syn-DBPDE of DBP, which is partially concord with wet-lab data.[13] ,[17] However, in-silico findings are consistent with GSTA1 against both carcinogenic metabolites, (-)- anti-DBPDE and (+)-syn-DBPDE of DBP
In addition, the binding pattern of GSTA1, M1, and P1 with week carcinogenic metabolites, (+)-antiDBPDE (GSTA1>M1>P1) and (-)-syn-DBPDE (GSTM2>A4>P1) is different and showed comparatively low activity. Furthermore, NAT2 activity is consistent and more than NAT1 either (±)-anti-DBPDE or (±)-syn-DBPDE metabolites of DBP, which is in accord with wet-lab findings.
No wet lab data investigated to date the direct involvement of NAT isozymes in the detoxification of (±)-anti-DBPDE and (±)-syn-DBPDE. However, its involvement in detoxification of certain monocyclic and heterocyclic amines is reported[33]-[35] and inferred that enzymatic activity of NAT2 appreciably more than NAT1.[33],[36] Our in-silico data exhibited preferable binding of NAT2 as compared to NAT1 against either (±)-anti-DBPDE or (±)-synDBPDE (Table 1). Thus, our computational findings are in reasonably good support to wet-lab data. Furthermore, this in-silico study provided additional insight into various GSTs and NATs against the DBP diol-epoxides over wet-lab experimentations.
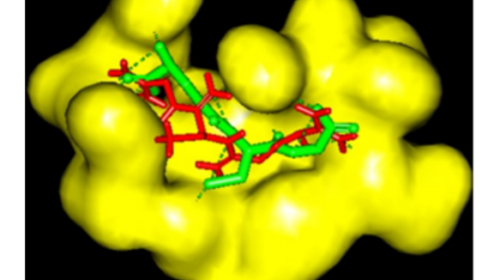
Figure 6: Validation of docking methodology through showing the same binding orientation of the ligand present within the active site of GSTP1 (green) and that after re-docking the same (red).
Validation of docking methodology
The ligand present in GSTP1 (PDB: 13GS) was extracted and dock within the active site using the same parameters as considered in (±)-anti-DBPDE and (±)-syn-DBPDE. The binding orientation of the ligand was the same as that in the parent molecule, confirming the accuracy of the binding and docking program used (Figure 6).
Metabolic detoxification of DMBADE with GST and NAT isozymes
Docking findings revealed that DMBADE showed plausible binding with GSTM1 (ΔG: -5.41 kcal/mol, Ki: 108.17 µM) followed by GSTP1 (ΔG: -5.29 kcal/mol, Ki: 133.38 µM) and GSTA1 (ΔG: -4.87 kcal/mol, Ki: 271.12 µM). Furthermore, PHE34, LYS33, LYS43, LEU44, ILE35, ASP40 residues of GSTM1, ARG70, ASN66, ARG13, SER65, GLU97, GLN64 residues of GSTP1, LYS138, and LYS141 residues of GSTA1 are involved in hydrophobic interaction of DMBADE (Figure 7a-c). The binding pattern of GSTA and GSTM isozymes against the DMBADE were followed the order as- GSTA1 >A3 >A2 >A4 >A5, GSTM1 >M2 >M4 >M5 > M3 (Table 2). In addition, our docking findings also revealed that NAT2 (ΔG: -8.06 kcal/mol, Ki: 1.24 µM) showed more favorable interactions than NAT1 (ΔG: -7.22 kcal/mol, Ki: 5.12 µM) with DMBADE (Table 2). VAL106, CYS68, SER125, PHE37, GLY126, PHE93, PHE217, THR289, and LEU288 residues of NAT2 are engaged hydrophobic interactions with DMBADE.

Figure 7: : Ligplot showing molecular interactions of DMBADE with a) GSTA1, b) GSTM1, c) GSTP1. Dashed lines and arcs show hydrophobic contacts and H-bonds, respectively

Table 2: Molecular docking studies of diol-epoxide of DMBA with GST and NAT isozymes.
Data published so far, emphasized that the carcinogenic metabolite of 7,12-DMBA, DMBA-3,4- diol-1,2-epoxide (DMBADE) is mainly conjugated by sulfotransferases.[37],[38], However, we have investigated GST and NAT isozymes in the conjugation of DMBADE instead of sulfotransferases. Our in-silico findings exhibited the feeble binding pattern among the different classes of GST as highlighted by their binding energies complementing the wet-lab data that the GSTs are not the primary detoxification enzyme for DMBADE.[38] In addition, no wet-lab data reveals the direct involvement of NAT isozymes in the detoxification of DMBADE. However, our insilico data showed preferable binding of NAT2 as compared to NAT1 against DMBADE. Thus, our computational findings highlighted the involvement of NAT isozymes while feebly support the conjugation by various GST isozymes.
Metabolic detoxification of N2-OH-PhIP with GST and NAT isozymes
The computational results revealed that N2-OHPhIP showed plausible binding with GSTP1 (ΔG: – 6.23 kcal/mol, Ki: 27.11 µM) followed by GSTM2 (ΔG: -5.76 kcal/mol, Ki: 59.86 µM) and GSTA3 (ΔG: -5.48 kcal/mol, Ki: 96.36 µM). Furthermore, GLN51, TYR49, GLN64, GLU97, ARG13, SER65, PRO53, and LEU52 residues of GSTP1 are involved in the hydrophobic interaction N2-OH-PhIP (Figure 8a-c). The binding pattern of GSTA and GSTM isozymes against the N2-OH-PhIP were followed the order asGSTA3 >A4>A5 >A1 >A2, GSTM2 >M3 >M4 >M1 > M5 (Table 3). In addition, docking simulation study also revealed that NAT2 (ΔG: -9.86 kcal/mol, Ki: 0.059 µM) showed more favorable interactions than NAT1 (ΔG: -8.56 kcal/mol, Ki: 0.530 µM) with N2-OH-PhIP (Table 3). SER287, LEU288, SER216, PHE217, CYS68, GLY124, HIS107, ASP122, PHE93, and PRO96 residues of NAT2 are engaged in hydrophobic interactions with N2-OH-PhIP.
The experimental data emphasizes glucuronidation of N2-OH-PhIP.[39],[40] , However, we have thoroughly examined the involvement of GST and NAT isozymes in the conjugation of N2-OH-PhIP. Findings exhibited the feeble binding pattern among the different classes of GST, as highlighted by their binding energies. In addition, our insilico data showed preferable binding of NAT2 compared to NAT1 against the N2-OH-PhIP. Thus, our computational findings highlighted the involvement of NAT isozymes while feebly support the conjugation by various GST isozymes.
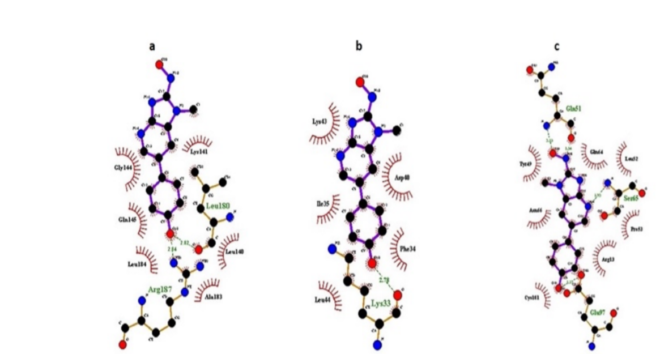
Figure 8: Molecular docking studies of diol-epoxide of DMBA with GST and NAT isozymes.

Table 3:: Molecular docking studies of N2-OH-PhIP with GST and NAT isozymes.
Metabolic detoxification of (±)-anti-BPDE, (±)-synBPDE with GST and NAT isozymes
The docking findings revealed that (-)-anti-PBDE showed plausible binding with GSTP1 (ΔG: -5.90 kcal/mol, Ki: 47.64 µM) followed by GSTM1 (ΔG: – 5.44 kcal/mol, Ki: 102.40 µM) and GSTA1 (ΔG: -4.26 kcal/mol, Ki: 753.46 µM). Furthermore, ARG13, GLN64, TYR49, GLN51, PRO53, LEU52 residues of GSTP1, LYS43, LEU44, ILE35, PHE34, LYS33, ASP40 residues of GSTM1, SER142, LYS138, LYS141, and PRO134 residues of GSTA1 (Figure 9a-c) are involved in hydrophobic interaction of (-)-antiBPDE. The binding pattern of GSTA and GSTM isozymes against the (-)-anti-BPDE were followed the order as- GSTA2>A4>A3>A1>A5, GSTM1>M4>M3>M2> M5 (Table 4). In addition, our docking findings also revealed that NAT2 (ΔG: – 7.19 kcal/mol, Ki: 5.34 µM) showed more favorable binding than NAT1 (ΔG: -6.98 kcal/mol, Ki: 29.75 µM) with (-)-anti-BPDE (Table 4). PHE150, ASP122, SER125, PHE93, TYR190, ILE189, GLN163, SER129, LEU135, and MET131 residues NAT2 are hydrophobically involved with (-)-anti-BPDE.
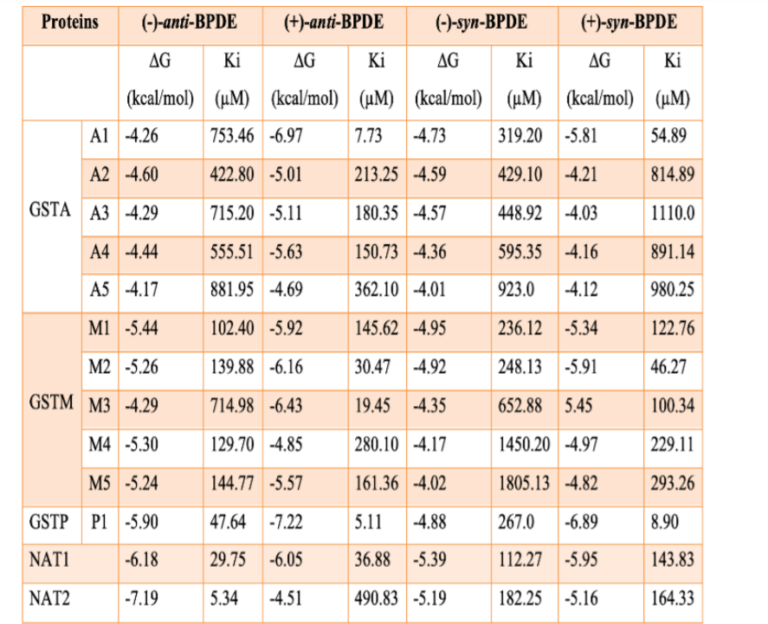
Table 4:Molecular docking studies of (±)-diolepoxides of BP with GST and NAT isozymes.
In continuation, (+)-anti-BPDE showed slightly more binding affinity and different order as-GSTP1 (ΔG: -7.22 kcal/mol, Ki: 5.11 µM) > GSTA1 (ΔG: – 6.97 kcal/mol, Ki: 7.73 µM) > GSTM3 (ΔG: -6.43 kcal/mol, Ki: 19.45 µM) when compared to (-)-antiBPDE. LYS141, LYS138 residues of GSTA1, TYR49, LEU44, LYS33, PHE34, LYS43 residues of GSTM1, ASN66, SER65, GLN64, ARG13, ILE104, and GLN51 residues of GSTP1 are engaged in hydrophobic interaction with (+)-anti-BPDE. The binding pattern of GSTA and GSTM isozymes against the (+)-anti-BPDE followed the order asGSTA1>A4>A3>A2>A5, GSTM3>M2>M1>M5>M4 (Table 4). In addition, our in-silico results also revealed that NAT1 (ΔG: -6.05 kcal/mol, Ki: 36.88 µM) showed more favorable binding than NAT2 (ΔG: -4.51 kcal/mol, Ki: 490.83 µM) with (+)-anti-BPDE (Table 4). ARG187, PHE125, ILE189, VAL93, ILE106, VAL216, PHE127, PHE287, and TYR94 residues of NAT1 are hydrophobically involved with (+)-anti-BPDE.
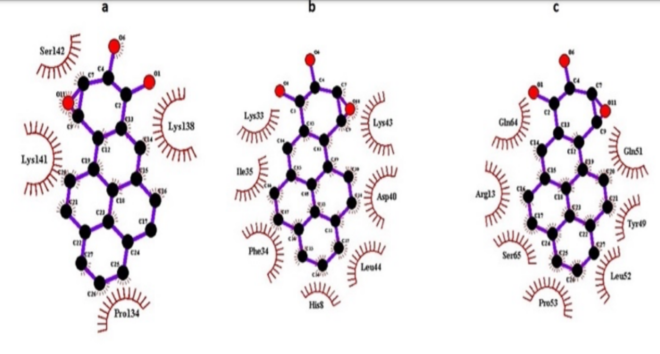
Figure 9: Ligplot showing molecular interactions of (-)-anti-BPDE with a) GSTA1, b) GSTM1, c) GSTP1. Arcs show hydrophobic contacts.
Our in-silico findings further elicited that (+)-synBPDE showed a more favorable binding affinity with GSTP1 (ΔG: -6.89 kcal/mol, Ki: 8.90 µM) followed by GSTM2 (ΔG: -5.91 kcal/mol, Ki: 46.27 µM) and GSTA1 (ΔG: -5.81 kcal/mol, Ki: 54.89 µM). LYS138, LYS141, GLU137, PRO134 residues of GSTA1, LYS43, LEU44, ILE35, PHE34, LYS33, ASP40 residues of GSTM1, GLN51, LEU52, PRO53, GLN64, ARG13, GLU97, and CYS101 residues of GSTP1 are involved in hydrophobic interaction of (+)-synBPDE. Furthermore, the binding pattern of GSTA and GSTM isozymes against the (+)-syn-BPDE followed the order as- GSTA1>A4>A2>A5>A3, GSTM2>M3>M1>M4>M5 (Table 4). In addition, our in-silico results also revealed that both NAT1 (ΔG: -5.95 kcal/mol, Ki: 143.83 µM) and NAT2 (ΔG: -5.16 kcal/mol, Ki: 166.33 µM) have almost identical binding against (+)-syn-BPDE (Table 4). PHE125, CYS68, HIS107, ILE106, PHE217, VAL216, PHE287, and VAL93 residues of NAT1 are hydrophobically involved with (+)-syn-BPDE. Further, (-)-syn-BPDE showed almost the same binding affinity as (+)-syn-BPDE with different GST and NAT isozymes having binding energy in the range of -4.01 to -5.39 kcal/mol (Table 4). The experimental data studied so far exhibited the more enzymatic activity of GSTM1 against the most potent diol-epoxide of BP, namely (+)-anti-BPDE and (+)-syn-BPDE followed by GSTP1 and GSTA1.[13],[31],[41], However, our in-silico findings showed the involvement of GSTs as GATP1>A1>M1, contrary to wet-lab data. Further, in-silico data exhibited preferable binding of NAT1 as compared to NAT2 against either (±)- anti-BPDE or (±)-syn-BPDE (Table 4). Thus, our computational findings are partially agreed with wet-lab data. However, these in-silico findings provided significant structural insight into various GST and NAT isozymes against the stereoisomeric diol-epoxides of BP over to wet-lab data.
Different isomeric forms of diol-epoxides of DBP, DMBA, PhIP, and BP have shown significant and more substantial molecular interactions with various isozymes of GST and NAT, suggesting that these metabolites could be highly reactive towards these enzymes. Subsequent interactions probably will lead to the inactivation of these detoxifying enzymes favoring the formation of DNA adducts from these diol-epoxides, which could lead to genetic instability. Our results may not be in complete agreement with the wet-lab findings for different carcinogenic metabolites due to several factors inside the cells and the constitutive actions of various metabolites together. Further, the simple fast-track protocol can be pretty valuable in predicting the GST and NAT isozymes involved in detoxifying the known metabolite(s) of PAHs.
This research did not received funding.
Authors like to acknowledge Integral University, Lucknow for providing necessary infra structure facilites to carry out this research.
References
- 1.Allegedly OO, Opeolu BO, Jackson VA. Polycyclic Aromatic Hydrocarbons: A Critical Review of Environmental Occurrence and Bioremediation. Environ Manage. 2017;60(4):758–83. [Google Scholar] [PubMed]
- 2.Abdel-Shafy HI, Mansour MSM. A review on polycyclic aromatic hydrocarbons: Source, environmental impact, effect on human health and remediation. Egypt J Pet. 2016;25(1):107– 23. [Google Scholar] [PubMed]
- 3.Shimada T, Fujii-Kuriyama Y. Metabolic activation of polycyclic aromatic hydrocarbons to carcinogens by cytochromes P450 1A1 and1B1. Cancer Sci. 2004;95(1):1–6. [Google Scholar] [PubMed]
- 4.Cavalieri EL, Rogan EG. Central role of radical cations in metabolic activation of polycyclic aromatic hydrocarbons. Xenobiotica. 1995;25(7):677–88 [Google Scholar] [PubMed]
- 5.Phillips DH, Grover PL. Polycyclic Hydrocarbon Activation: Bay Regions and Beyond. Drug Metab Rev. 1994;26(1–2):443–67. [Google Scholar] [PubMed]
- 6.Jernström B, Gräslund A. Covalent binding of benzo[a]pyrene 7,8-dihydrodiol 9,10-epoxides to DNA: molecular structures, induced mutations and biological consequences. Biophys Chem. 1994;49(3):185–99. [Google Scholar] [PubMed]
- 7.Khan MKA, Salman A, Al-Khodairy Salman F, AlMarshad Feras M, Alshahrani Abdulrahman M, Arif Jamal M. Computational Exploration of Dibenzo[a,l]Pyrene Interaction to DNA and its Bases: Possible Implications to Human Health. Biointerface Res Appl Chem. 2020;11(4):11272–83. [Google Scholar] [PubMed]
- 8.Sar D, Kim B, Ostadhossein F, Misra SK, Pan D. Revisiting Polyarenes and Related Molecules: An Update of Synthetic Approaches and Structure-Activity-Mechanistic Correlation for Carcinogenesis. Chem Rec. 2018;18(6):619–58. [Google Scholar] [PubMed]
- 9.Lin Y-C, Chou F-C, Li Y-C, Jhang S-R, Shangdiar S. Effect of air pollutants and toxic emissions from various mileage of motorcycles and aerosol related carcinogenicity and mutagenicity assessment. J Hazard Mater. 2019; 365:771–7. [Google Scholar] [PubMed]
- 10.Shimada T. Xenobiotic-Metabolizing Enzymes Involved in Activation and Detoxification of Carcinogenic Polycyclic Aromatic Hydrocarbons. Drug Metab Pharmacokinet. 2006;21(4):257–76. [Google Scholar] [PubMed]
- 11.Sayer JM, Yagi H, van Bladeren PJ, Levin W, Jerina DM. Stereoselectivity of microsomal epoxide hydrolase toward diol epoxides and tetrahydroepoxides derived from benz[a]anthracene. J Biol Chem. 1985;260(3):1630–40 [Google Scholar] [PubMed]
- 12.Hecht SS, Carmella SG, Villalta PW, Hochalter JB. Analysis of Phenanthrene and Benzo[a]pyrene Tetraol Enantiomers in Human Urine: Relevance to the Bay Region Diol Epoxide Hypothesis of Benzo[a]pyrene Carcinogenesis and to Biomarker Studies. Chem Res Toxicol. 2010;23(5):900–8. [Google Scholar] [PubMed]
- 13.Jernström B, Funk M, Frank H, Mannervik B, Seidel A. CARCINOGENESIS: Glutathione Stransferase A1–1-catalysed conjugation of bay and fjord region diol epoxides of polycyclic aromatic hydrocarbons with glutathione. Carcinogenesis. 1996;17(7):1491–8. [Google Scholar] [PubMed]
- 14.Romert L, Dock L, Jenssen D, Jernström B. Effects of glutathione transferase activity on benzo[a]pyrene 7,8-dihydrodiol metabolism and mutagenesis studied in a mammalian cell co-cultivation assay. Carcinogenesis. 1989;10(9):1701–7. [Google Scholar] [PubMed]
- 15.Hesse S, Cumpelik O, Mezger M, Kiefer F, Wiebel FJ. Glutathione conjugation protects some, but not all, cell lines against DNA binding of benzo[a]pyrene metabolites. Carcinogenesis. 1990;11(3):485–7 [Google Scholar] [PubMed]
- 16.Nerurkar P V., Okinaka L, Aoki C, Seifried A, Lum-Jones A, Wilkens LR, et al. CYP1A1, GSTM1, and GSTP1 genetic polymorphisms and urinary 1-hydroxypyrene excretion in nonoccupationally exposed individuals. Cancer Epidemiol Biomarkers Prev. 2000;9(10):1119- 22. [Google Scholar] [PubMed]
- 17.Sundberg K, Widersten M, Seidel A, Mannervik B, Jernström B. Glutathione Conjugation of Bayand Fjord-Region Diol Epoxides of Polycyclic Aromatic Hydrocarbons by Glutathione Transferases M1-1 and P1-1. Chem Res Toxicol. 1997;10(11):1221–7. [Google Scholar] [PubMed]
- 18.Kabir S, Rehman A. Carcinogenic potential of arylamine N-acetyltransferase in Asian populations. J Cancer Res Pract. 2018;5(4):131– 5 [Google Scholar] [PubMed]
- 19.Doll MA, Zang Y, Moeller T, Hein DW. Codominant Expression of N -Acetylation and O Acetylation Activities Catalyzed by N – Acetyltransferase 2 in Human Hepatocytes. J Pharmacol Exp Ther. 2010;334(2):540–4. [Google Scholar] [PubMed]
- 20.Hein DW, Millner LM. Arylamine Nacetyltransferase acetylation polymorphisms: paradigm for pharmacogenomic-guided therapy- a focused review. Expert Opin Drug Metab Toxicol. 2021;17(1):9–21. [Google Scholar] [PubMed]
- 21.Hein DW. N-Acetyltransferase genetics and their role in predisposition to aromatic and heterocyclic amine-induced carcinogenesis. Toxicol Lett. 2000;112–113:349–56. [Google Scholar] [PubMed]
- 22.Moore LE, Baris DR, Figueroa JD, Garcia-Closas M, Karagas MR, Schwenn MR, et al. GSTM1 null and NAT2 slow acetylation genotypes, smoking intensity and bladder cancer risk: results from the New England bladder cancer study and NAT2 meta-analysis. Carcinogenesis. 2011;32(2):182–9. [Google Scholar] [PubMed]
- 23.Sim E, Abuhammad A, Ryan A. Arylamine Nacetyltransferases: from drug metabolism and pharmacogenetics to drug discovery. Br J Pharmacol. 2014;171(11):2705–25. [Google Scholar] [PubMed]
- 24.Khan MKA, Akhtar S, Arif JM. Development of In Silico Protocols to Predict Structural Insights into the Metabolic Activation Pathways of Xenobiotics. Interdiscip Sci Comput Life Sci. 2018;10(2):329–45. [Google Scholar] [PubMed]
- 25.Rehman A, Akhtar S, Siddiqui MH, Sayeed U, Ahmad SS, Arif JM, Khan MKA. Identification of potential leads against 4- hydroxytetrahydrodipicolinate synthase from Mycobacterium tuberculosis. Bioinformation. 2016;12(11):400–7. [Google Scholar] [PubMed]
- 26.Prymula K, Jadczyk T, Roterman I. Catalytic residues in hydrolases: analysis of methods designed for ligand-binding site prediction. J Comput Aided Mol Des. 2011;25(2):117–33. [Google Scholar] [PubMed]
- 27.Huey R, M. Morris G, Olson AJ, Goodsell DS. A Semiempirical Free Energy Force Field with Charge-Based Desolvation. J Comput Chem. 2007;28(6):1145–52. [Google Scholar] [PubMed]
- 28.Laskowski RA, Swindells MB. LigPlot+: Multiple Ligand–Protein Interaction Diagrams for Drug Discovery. J Chem Inf Model. 2011;51(10):2778–86. [Google Scholar] [PubMed]
- 29.Hu X. Specificities of human glutathione Stransferase isozymes toward anti- diol epoxides of methylchrysenes. Carcinogenesis. 1998;19(9):1685–9. [Google Scholar] [PubMed]
- 30.Xia H, Pan S-S, Hu X, Srivastava SK, Pal A, Singh S V. Cloning, Expression, and Biochemical Characterization of a Functionally Novel Alpha Class GlutathioneS-Transferase with Exceptional Activity in the Glutathione Conjugation of (+)-Anti-7,8-dihydroxy-9,10- oxy-7,8,9,10-tetrahydrobenzo(a)pyrene. Arch Biochem Biophys. 1998 ;353(2):337–48. [Google Scholar] [PubMed]
- 31.Sundberg K, Dreij K, Seidel A, Jernström B. Glutathione Conjugation and DNA Adduct Formation of Dibenzo[a,l]pyrene and Benzo[a]pyrene Diol Epoxides in V79 Cells Stably Expressing Different Human Glutathione Transferases. Chem Res Toxicol. 2002;15(2):170–9. [Google Scholar] [PubMed]
- 32.Axarli I, Muleta AW, Chronopoulou EG, Papageorgiou AC, Labrou NE. Directed evolution of glutathione transferases towards a selective glutathione-binding site and improved oxidative stability. Biochim Biophys Acta-Gen Subj. 2017;1861(1):3416–28. [Google Scholar] [PubMed]
- 33.Mitchell SC. N-acetyltransferase: the practical consequences of polymorphic activity in man. Xenobiotica. 2020;50(1):77–91. [Google Scholar] [PubMed]
- 34.Delannée V, Langouët S, Siegel A, Théret N. In silico prediction of Heterocyclic Aromatic Amines metabolism susceptible to form DNA adducts in humans. Toxicol Lett. 2019;300:18– 30. [Google Scholar] [PubMed]
- 35.Uno Y, Murayama N, Yamazaki H. Genetic variants of N-acetyltransferases 1 and 2 (NAT1 and NAT2) in cynomolgus and rhesus macaques. Biochem Pharmacol. 2020;177:113996. [Google Scholar] [PubMed]
- 36.Laurieri N, Sim E. Arylamine N – Acetyltransferases in Health and Disease. Arylamine N -Acetyltransferases in Health and Disease. WORLD SCIENTIFIC; 2018. [Google Scholar] [PubMed]
- 37.Sarah Abdelrahman Mohamed, Ayman Saber Mohamed, Emad El-Zayat, Mohamed Refaat Shehata. Protective and curative mechanisms of echinochrome against 7,12- Dimethylbenz[a]anthracene -induced renal toxicity in rats. GSC Adv Res Rev. 2021;6(1):047–55. [Google Scholar] [PubMed]
- 38.Nagah A, Amer A. Different Mechanisms of Cigarette Smoking-Induced Lung Cancer. Acta Biotheor. 2021;69(1):37–52 [Google Scholar] [PubMed]
- 39.Dellinger RW, Chen G, Blevins-Primeau AS, Krzeminski J, Amin S, Lazarus P. Glucuronidation of PhIP and N-OH-PhIP by UDP-glucuronosyltransferase 1A10. Carcinogenesis. 2007;28(11):2412–8. [Google Scholar] [PubMed]
- 40.Malfatti MA, Ubick EA, Felton JS. The impact of glucuronidation on the bioactivation and DNA adduction of the cooked-food carcinogen 2- amino-1-methyl-6-phenylimidazo[4,5- b]pyridine in vivo. Carcinogenesis. 2005;26(11):2019–28. [Google Scholar] [PubMed]
- 41.Sundberg K. Differences in the catalytic efficiencies of allelic variants of glutathione transferase P1-1 towards carcinogenic diol epoxides of polycyclic aromatic hydrocarbons. Carcinogenesis. 1998;19(3):433–6. [Google Scholar] [PubMed]